探索作用于无药可肿瘤靶点RAS的下游通路的抑制技术以及可用于测试RAS靶标药物的临床前模型。
RAS家族突变和癌症
每年确诊的癌症中有三分之一是由RAS家族基因突变引起的,包括95%的胰腺癌和45%的结肠癌。理论上,这为治疗多种癌症提供了一系列有吸引力的靶点。
RAS家族的三名成员包括:
- KRAS是最常见的突变(在所有RAS驱动的癌症中占85%)
- 其次是NRAS(占12%).
- HRAS (占3%).
这三种RAS蛋白共享超过80%的氨基酸序列,突变主要发生在密码子12,13和61这三个基因中。
RAS蛋白和细胞增殖
RAS是膜相关的G蛋白,这意味这他们可以结合GTP和GDP。当RAS与GTP结合时,该蛋白被转换成激活状态从而诱导细胞生长,增殖和迁移。RAS与GDP结合则会转换成非激活状态。当RAS家族成员发生突变时,他们会永久地与GTP结合从而不断促进细胞增殖。
RAS:一种无药可肿瘤靶点
有一种阻止致癌RAS的方案是阻断其激活。然而,开发针对突变RAS蛋白的药物的尝试一直没有成功。这是由于GTP的相对细胞丰度以及RAS结合GTP的亲和力极高造成的。 同时也是由于在RAS蛋白的关键区域明显缺乏与小分子结合所必须的合适表面,这使得携带这些突变的肿瘤成为了最难治疗的肿瘤之一。目前与RAS相关的治疗策略是使用一种间接方法,转而针对RAS的下游通路。
RAS下游通路药物靶点
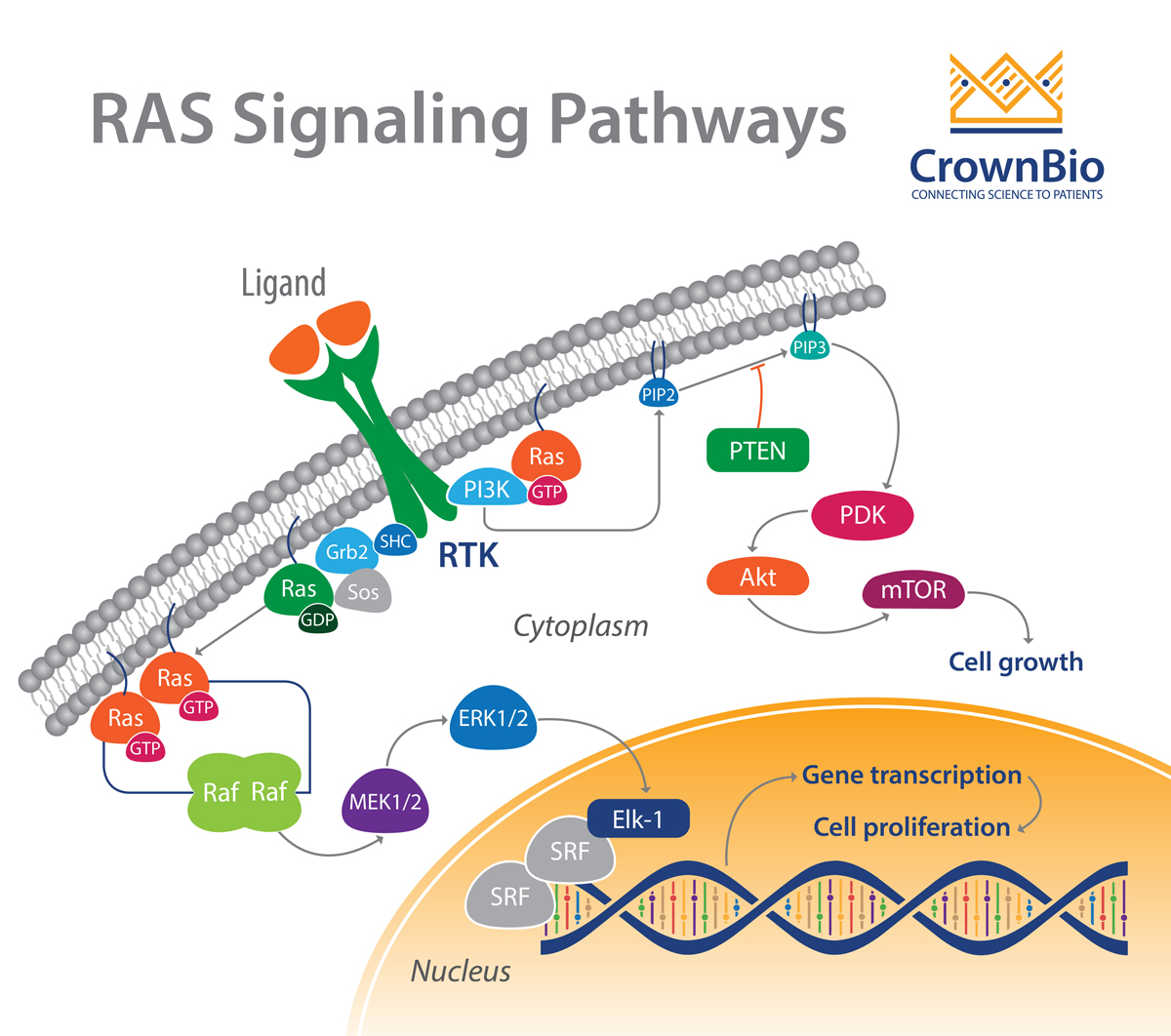
RAS-RAF-M3K-ERK
RAS-RAF-MEK-ERK(MAPK)级联是由配体与受体酪氨酸激酶(RTK)结合而引发的一种重要的信号通路。一旦RAF被激活,RAF磷酸化并激活丝裂原活化蛋白激酶1和2(MEK1和MEK2)。这些蛋白可以同时激活下游细胞外信号调节激酶1和2(ERK1和ERK2)。
ERK1和ERK2通过磷酸化来调控细胞核中的转录因子(比如c-Myc,c-Fos和CREB),作用于基因表达,参与细胞生长,分化和分裂。
P13K-Akt
RAS还与PI3K-Akt通路相互作用并刺激PI3K-Akt通路。一旦PI3K被RAS激活,AKT就会被磷酸化并被激活,产生一系列的下游效应,包括激活mTOR来调节细胞生长、增殖和血管生成所必需的蛋白质的合成。
目前的抑制途径:BRAF和MEK
使用BRAF抑制剂(如vemurafenib、dabrafenib)或MEK抑制剂(如trametinib, cobimetinib)靶向MAPK信号已被证明是治疗多种不同癌症的有效方法。然而,耐药性仍然是一个主要的挑战——30%的肿瘤对抑制剂没有反应,70%有反应的肿瘤通常能看到进展。
RAS靶向治疗的临床前模型
多种临床前模型可用于评估靶向RAS及其下游通路的药物,包括基因工程小鼠模型(GEMM)和人源性异种移植模型(PDX)。
基因工程小鼠模型
临床前GEMM 概括了与肿瘤进展和发展密切相关的基因被删除、过度表达或突变的疾病特征。这导致自发性肿瘤的形成。
目前有大量的GEMM可用,包括许多常见的癌症适应症,如肺癌、前列腺癌、乳腺癌、结肠癌和胰腺癌。
KRAS突变GEMM测试针对RAS癌基因家族的治疗方法特别有趣。事实上,KRAS突变在肺、胰腺和胃肠道的条件表达可诱导癌前上皮增生、腺瘤、胰腺上皮内瘤变和腺癌。这些模型已成功地用于证明BRAF、MEK和mTOR抑制剂作为单药和联合用药的有效性。
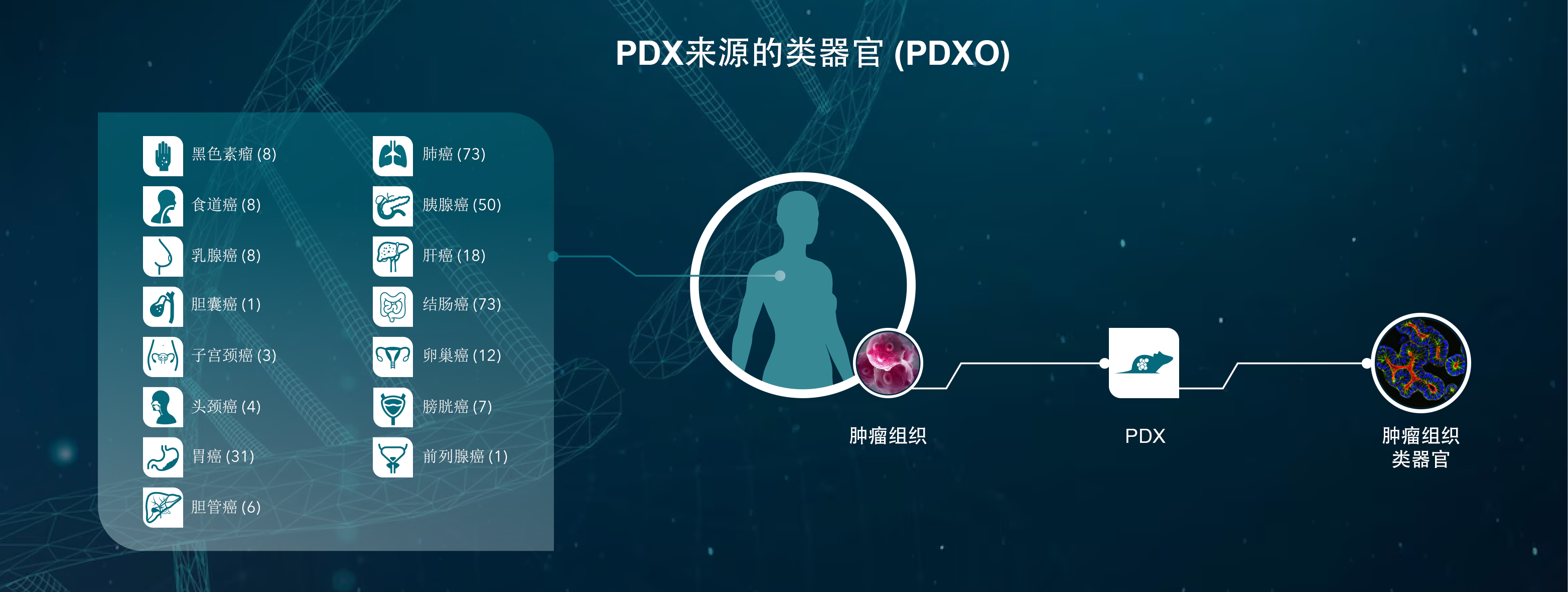
人源性肿瘤异种移植模型
PDX也是评估RAS靶向治疗的有价值的转化临床前模型。PDX是直接从患者组织样本中提取的动物模型,它保持着从患者身上提取的基因型和表型的保真度。这比传统的异种移植提供了更多的预测模型。
PDX的时间跟病人接近,从来没有受到细胞培养选择的压力,更紧密地概括病人疾病。这些模型不仅在肿瘤的组织学表现上显示出很高的保真度,而且在对化疗、放疗和靶向治疗的反应上也同样如此。
携带相关突变的PDX模型已成功地用于评估RAS下游靶点抑制剂的有效性,并在最近被用于鉴定小分子化合物与KRAS的结合。
总结
RAS蛋白是控制细胞生长、增殖和迁移的信号网的重要组成部分。RAS基因突变经常在人类肿瘤中发现,并且对靶向治疗非常抵触。
概括了人类疾病的关键方面GWMM和PDX RAS突变模型,为肿瘤学研究提供了重要模型,并被广泛用于靶点和治疗方法的临床前验证。