探索以各种细胞DNA损伤应答(DDR)和DNA修复通路为靶点开发新抗癌治疗策略的途径
癌症的DNA损伤应答和DNA修复
发生癌症时,DNA修复和DNA损伤应答(DDR)通路经常遭到破坏,这是癌症的标志之一。关键DNA修复/DDR基因的种系和/或体细胞突变分别导致对癌症的易感性和在癌症中出现较高的突变负担。
合成致死是一个基因组概念,即通常在同一通路中同时破坏两个基因,导致细胞死亡。重要的是,合成致死已被证明发生在基因与药物之间,这一方法已成功地应用于携带DNA修复/DDR缺陷的肿瘤。批准将聚(ADP-核糖)聚合酶(PARP)抑制剂用于治疗BRCA1/2突变的卵巢癌就是例证。在这种情况下,一个基因因突变而失活,另一个因药物而失活。
DNA修复过程
细胞不断地应对因内源性过程(如DNA复制压力)或外源性暴露(如电离辐射和化疗药物)造成的DNA损伤。DNA损伤剂会导致不同类型的DNA损伤,而无法修复损伤会对细胞造成一系列可能的毁灭性后果,包括基因不稳定性和促进肿瘤形成的突变积累。因此,细胞形成了复杂的修复机制,来处理可能出现的不同类型DNA损伤;所有这些都是为了保护基因组的稳定性。
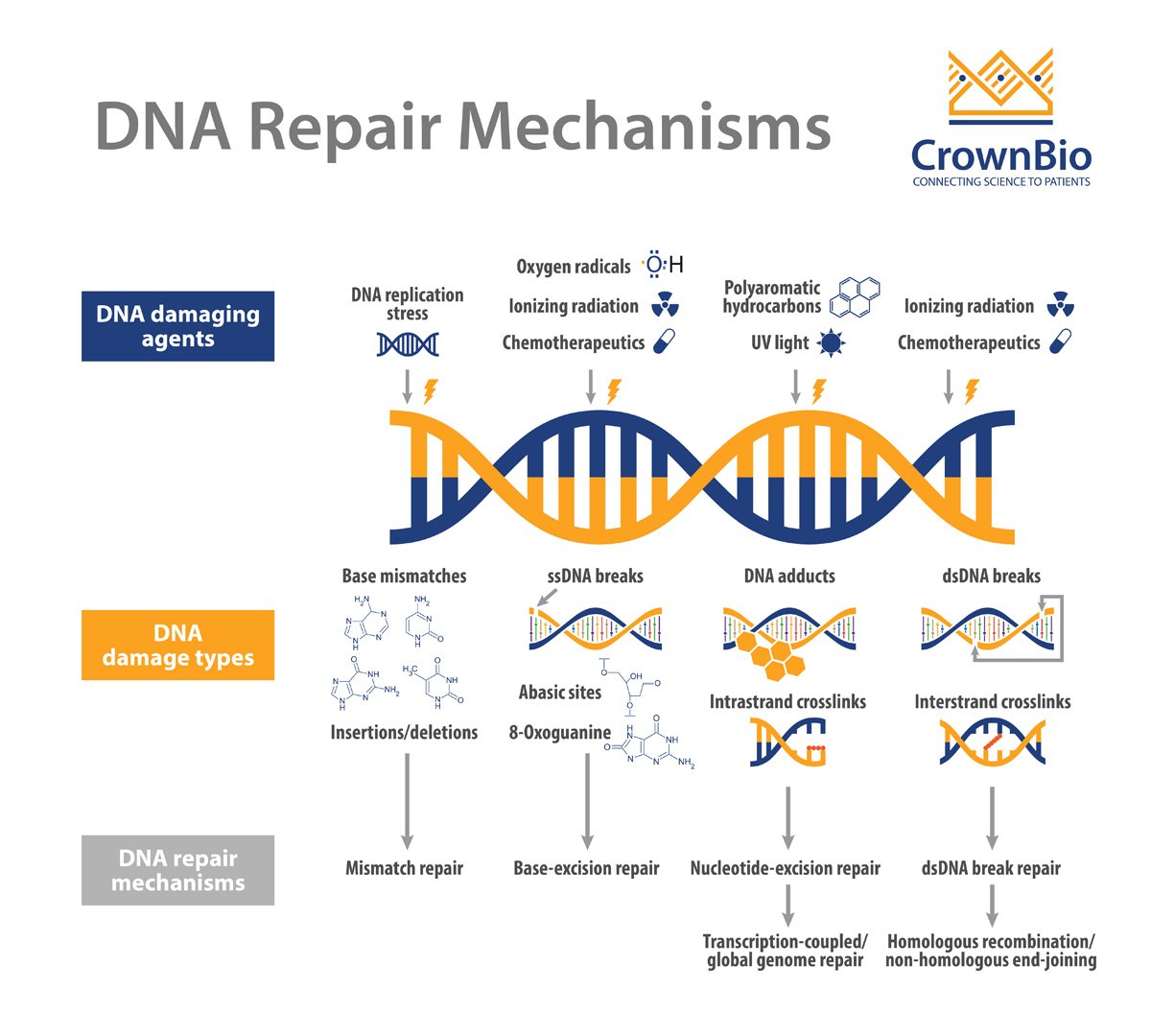
DNA修复机制的主要类型包括:
直接修复
直接修复是最简单的DNA修复形式,因为它主要依赖于单个蛋白的活性,且无需进行核苷酸去除、重新合成或结扎。例如,O6-甲基鸟嘌呤(O6-mG)是一种由烷基化诱变剂引起的有害基因损伤。鸟嘌呤O6位点存在烷基导致G:C转变至A:T和基因转录或DNA复制阻滞。在直接修复过程中,烷基被O6-甲基鸟嘌呤DNA甲基转移酶(MGMT)去除,适当的核苷酸被恢复。
碱基切除修复(BER)
BER负责修复对基因组保真度和稳定性具有重大威胁的较小但高度诱变DNA损伤。这些DNA损伤可由电离辐射以及氧化、甲基化、脱氨等代谢活动或DNA碱基对的自发丢失所产生的内源性诱变引起。BER通路由11种取出受损碱基的DNA糖苷酶之一启动。碱基切除后,一组不同的蛋白通过单个碱基(短补丁通路)或多个碱基(长补丁通路)填充暴露的间隙。
核苷酸切除修复(NER)
就已经识别和修复损伤的多样性而言,NER可能是最灵活的机制。在这些损伤中,最显著的是UV光诱导的嘧啶二聚体(环丁烷嘧啶二聚体和6-4光产物),其他NER底物包括大体积化学加合物、DNA链间交联和某些形式的氧化损伤。这些类型的损伤会导致DNA双链螺旋扭曲和DNA化学改变,这两者都是NER识别的标志性特征。
NER是一个复杂的多步骤过程,涉及一个由30多种蛋白组成的大网络。首先识别受损碱基,然后解开DNA双链,并通过切除修复复合物去除受损碱基,然后进行填充和结扎。
DNA错配修复(MMR)
MMR通路在修正复制错误中起着至关重要的作用,例如分别由核苷酸的DNA聚合酶错掺和模板滑移导致的碱基-碱基错配和插入/缺失环(IDL)。MMR还纠正了由5-甲基胞嘧啶的自发脱氨基作用和基因重组后形成的异源双链核酸分子所产生的错配对或“错配”。
该通路的缺陷导致一种“突变体”细胞表现型,其特征是自发突变频率升高和微卫星不稳定性(MSI)增加。几种人类MMR基因的突变导致对遗传性非息肉病性结直肠癌(HNPCC)的易感性,以及多种表现MSI的散发性肿瘤。
双链断裂(DSB)修复
DSB是一种高毒性的基因损伤,对细胞自动调节构成严重威胁,因为它们可以影响转录、复制和染色体分离。DSB由多种外源性因素引起,如电离辐射和某些基因毒性化学物质,以及内源性因素,如活性氧簇、单链DNA断裂的复制和染色体上的机械应力。
DSB不同于大多数其他类型的DNA损伤,主要是因为它们影响DNA的双链,因此阻止使用互补链作为模板进行修复(即BER、NER和MMR)。无法修复DSB可导致毁灭性的染色体不稳定性,从而导致基因表达异常和致癌风险增加。
细胞已经进化出两种不同的DSB修复通路:同源重组(HR)和非同源性末端接合(NHEJ)。虽然细胞可能选择使用其中的任何一种通路来修复DSB,但细胞选择其中一种通路而非另一种通路的原因仍然不明。在损伤发生时,选择似乎受到细胞周期阶段的影响。
HR
HR通过修复DSB、间隙和重启停滞的复制叉来维持基因组的稳定性。这是一种相对缓慢但无错误的通路,它依赖于基因组中存在的同源序列作为模板来替换受损的DNA片段。
NHEJ
与HR不同,NHEJ不需要使用DNA模板(姊妹染色单体)进行修复。相反,NHEJ通过使用不同的核酸酶修饰断裂任意一侧的DNA自由端,使其变得具有兼容性(即3′-羟基和5′-磷酸盐),然后与DNA连接酶4连接。与HR相比,NHEJ是一个相对快速但本质上容易出错的过程,过度使用可导致基因重排、缺失和突变,所有这些均可导致复制后细胞更易受到DSB的伤害。
DNA损伤感受器蛋白和DNA损伤信号蛋白可作为一系列抗癌药物的靶点。
DNA损伤感受器蛋白的药物靶向治疗
DDR对修复通路的激活和细胞存活至关重要,而对多种DNA损伤做出应答的DDR感受器蛋白是启动修复的关键。
对于DSB,Ku和MRN是主要感受器蛋白复合物。Ku是一种由Ku70/Ku80构成的蛋白异质二聚体,是NHEJ机制的一部分,能立即与DNA DSB结合。在识别和结合DSB后,Ku吸收其他蛋白,以协助进行经典NHEJ修复。
MRN复合物对DSB的初始检测也很重要。在与受损位点结合后,MRN吸收DNA损伤信号激酶ATM,激活并触发一系列信号事件,启动DNA末端切除并促进HR修复。已鉴别出多种其他DNA损伤感受器,包括范可尼贫血核心复合物、错配修复蛋白和核苷酸切除修复蛋白,它们是DNA链间交联、碱基-碱基错配或插入-缺失环以及UV诱导光损伤的感受器。
PARP-1是一种关键DNA损伤感受器蛋白。由于PARP与BRCA之间存在合成致死关系,已在很大程度上将它作为药物靶点。当同时丧失两个基因的功能(通常在同一通路中)导致细胞死亡时,就会出现合成致死。通过小分子抑制剂(如奥拉帕尼、尼拉帕利、尼拉帕利和他拉唑帕尼)在BRCA突变卵巢癌中成功实现临床PARP靶向治疗,提供了利用合成致死作为治疗策略的原理验证,并已对以DNA损伤应答为靶点的其他疗法进行开发。
DNA损伤信号蛋白的药物靶向治疗
DDR信号蛋白触发各种使DNA损伤信号放大和多样化的翻译后修饰和蛋白复合物装配,以便能够启动适当的应答,并且能够包括:转录变化、细胞周期检查点激活、选择性剪接、参与DNA修复过程,或在大规模损伤的背景下,激活细胞衰老和凋亡通路。
下文将讨论协调DDR信号事件与以此通路为靶点的新药物的主要蛋白。
DNA-PK
NHEJ正常修复DSB需要DNA蛋白激酶(DNA- pk),是贯穿细胞周期各个阶段人类细胞中DSB的主要DNA修复通路。DNA-PK激酶活性的小分子抑制剂使DNA-PK在DNA末端稳定,随后损害NHEJ,还可能干扰其他修复过程,包括通过阻碍DNA末端切除实现的HR。DNA-PK活性的丧失导致细胞增殖减少和胱天蛋白酶介导的细胞死亡启动。
由于与其他激酶的结构相似,与靶向DNA-PK相关的挑战之一是选择性。鉴于它在NHEJ中的作用,经发现在与诱导不依赖复制DSB的药物,如电离辐射和拓扑异构酶2抑制剂(如阿霉素和依托泊苷),联合使用时,以DNA-PK为靶点的药物更为有效。
一些小分子目前正处于不同的临床开发阶段,包括MSC2490484A、VX-984和CC-115。
ATM
ATM是一种在整个细胞周期中促进DSB修复和响应DSB蛋白激酶。ATM主要通过与MRN复合物的NBS1相互作用激活。它是负责组蛋白H2AX磷酸化的主要激酶,在DSB后迅速发生,并作为DNA修复机制装配的基础。经证明,ATM抑制使细胞对电离辐射和DSB药物(如依托泊苷、喜树碱和阿霉素)非常敏感。
目前,正在评价ATM抑制剂AZD0156的一期试验,作为单药治疗和与PARP抑制剂奥拉帕尼以及其他细胞毒类药物(包括伊立替康)联合使用。
ATR
在HR的早期阶段,ATR被ssDNA位点上结合的复制蛋白A (RPA),如停滞的复制叉,或随后在其中一条DNA链发生的5′-3’降解(即DNA末端切除)激活。
Berzosertib(也被称为M6620和VX-970)是首创ATR抑制剂,临床前数据显示肺癌细胞主要对化疗药物敏感。这导致了复制叉崩溃,如顺铂和吉西他滨(体外),并在与顺铂(体内)联合时提高了抗肿瘤活性。
CHK1
CHK1是ATR下游的蛋白激酶,是S和G2-M细胞周期检查点的关键调节因子。考虑到CHK1在DNA损伤后介导细胞周期阻滞中的作用,当与在DNA复制过程中诱导DNA损伤的药物联合使用时,CHK1抑制剂似乎最为有效,也就不足为奇了。因此,它们的临床开发集中在与此类药物的联合使用上。
MK8776是一种有效的选择性CHK1抑制剂。经证明,单药治疗和与吉西他滨联合使用均具有良好的耐受性。最近,在临床前和临床试验中,CHK1抑制剂prexasertib也显示出具有单药和联合活性。
WEE1
与CHK1平行作用的WEE1蛋白激酶,通过调节细胞周期蛋白依赖性激酶在激活G2-M检查点中发挥重要作用。但是,与CHK1不同的是,WEE1不直接受DNA损伤的调节,但它是生理细胞周期进程所必需的蛋白激酶。
对于WEE1抑制剂,我们认为其作用机制是通过阻止作为不适当CDK1/CCNB1激活结果的G2-M检查点的激活,从而导致有丝分裂灾变。但是,近期的数据表明,由于CDK2抑制导致DNA复制异常,WEE1抑制也会导致细胞中出现复制依赖性DNA损伤。
经证明,首创WEE1激酶抑制剂AZD1775可增加一系列DNA损伤剂的细胞毒性作用,并已在临床前模型中证明了单药活性。最近,一项一期研究发现,AZD1775与拓扑异构酶抑制剂联合使用具有可耐受性,目前已转入二期研究。
虽然NHEJ和HR是主要的DSB修复通路,但选择性同源重组修复机制的重要性正日益得到公认。例如,已知HR缺乏细胞依赖容易出错的微同源介导末端连接(MMEJ)来进行DNA修复和存活。在MMEJ过程中,间隙填充需要POLQ的DNA聚合酶活性(DNA聚合酶θ),且POLQ还通过限制RAD51在切除的DNA末端的积累来防止过度重组。因此,POLQ是一个具有吸引力的药物靶点,尤其在HR缺乏肿瘤中。
正在开发破坏RAD51重组酶家族蛋白-蛋白相互作用的小分子抑制剂,可能对HR缺乏肿瘤特别有效。在近期的一项分析中,确定了14种正处于临床开发中且以DDR为靶点的化合物(除PARP1/2抑制剂以外),并在临床前环境中评价了其他药物。
DNA损伤应答(DDR)联合策略
在许多类型的癌症中,有效应对DNA损伤的能力往往会丧失,以这一通路为靶点的药物已在患者亚群中得到了临床验证,如使用PARP抑制剂治疗BRCA1/BRCA2突变的患者。为了提高治疗效果,DDR抑制剂可与以其他DDR蛋白或完全不同的信号通路为靶点的药物联合使用,目的是阻断癌细胞赖以生存的多种通路。
例如,目前临床试验正在检测PARP抑制剂与以DRR通路的其他成员为靶点的小分子的联合使用,包括AZD6738 (ATR)、AZD0156 (ATM)或AZD1775 (WEE1),以及ATM缺乏肿瘤中的ATR抑制、细胞周期蛋白E或MYC扩增肿瘤中的靶向WEE1和HRD或NHEJ缺乏肿瘤中的POLQ抑制剂。临床前研究还表明,以DDR蛋白为靶点对治疗癌基因表达失调的肿瘤可能有利,因为强致癌信号可诱导复制压力。
DDR抑制剂也可与标准治疗药物联合使用,如使用PARP抑制剂增强铂类药物的效果,以及评价其他DDR抑制剂(包括CHK1/2和WEE1抑制剂)与放化疗联合使用的其他研究。
最后,存在重要的科学依据和临床证据表明,DDR和免疫应答相互关联,且具有潜在协同性。随着我们对DNA损伤、DDR与免疫应答之间相互作用理解的不断加深,将其与DDR抑制剂和/或辐射(作为增敏剂)联合使用可能会提高免疫疗法的临床疗效。
例如,当基因损伤未修复时,会导致肿瘤细胞表面突变负荷和新抗原表达的巨大改变。临床前研究表明,在患BRCA突变型卵巢癌的小鼠模型中,联合使用DNA修复靶向治疗与免疫检查点抑制剂可产生协同效应,如双重CTLA-4和PARP阻断,从而降低肿瘤负荷并提高生存率。
在临床环境中,早期研究支持联合使用PARP抑制剂与抗PD/PD-L1药物的安全性20。目前正在进行多项试验,探索与其他DDR抑制剂的联合使用,包括以ATR为靶点的DDR抑制剂。
结论
虽然癌症细胞可能受益于DDR通路中的缺陷,但存在其他起作用的DNA修复系统可提高存活率。以DDR为靶点的药物已在患者亚群中得到了临床验证,且正在积极研究不同的组合策略,以抑制癌细胞生存所依赖的多个通路。通过阻碍DNA修复,DDR抑制剂是适用于联合治疗的理想药物,可以提高辐射、化疗和免疫治疗的疗效。
延伸阅读
Brown et al. Targeting DNA repair in cancer: Beyond PARP inhibitors. Cancer Discovery 2017;7: 20–37.