 探索“不可成药”的肿瘤靶点 RAS、用于下游通路的抑制技术,及用于测试新兴 RAS 靶向药剂的临床前模型。
探索“不可成药”的肿瘤靶点 RAS、用于下游通路的抑制技术,及用于测试新兴 RAS 靶向药剂的临床前模型。
RAS家族突变与癌症
每年确诊的癌症中有三分之一是由 RAS 家族基因的突变引起的,其中包括95%的胰腺癌和45%的结直肠癌。从理论上讲,这为治疗多种癌症类型提供了一个有吸引力的靶点家族。
RAS家族由三位成员代表:
- KRAS,突变频率最高(在所有由 RAS 引起的癌症中占比85%)。
- 其次是 NRAS(12%)。
- 最后是 HRAS(3%)。
这三种 RAS 蛋白在氨基酸序列上有超过80%的相似性,突变主要发生在三个基因的第 12、13 和 61 个密码子上。
RAS 蛋白与细胞增殖
RAS 蛋白是与细胞膜相关的G蛋白,这意味着它们能够结合 GTP 和 GDP。当 RAS 与 GTP 结合时,蛋白转变为活性状态,诱导细胞生长、增殖和迁移。结合 GDP 则使蛋白转变为非活性状态。
当 RAS 家族成员发生突变时,它们会永久性地结合到 GTP 上,持续地推动细胞增殖。
RAS:一个“不可成药”的肿瘤学靶点
阻止致癌 RAS 的一个解决办法是阻止其激活。然而,尝试开发针对突变 RAS 蛋白的药物一直未能成功。
这是由于 GTP 在细胞中的相对丰度以及RAS对GTP的结合亲和力极高。同时,在 RAS 蛋白的关键区域显然缺乏适合小分子结合的合适表面,这使得携带这些突变的肿瘤成为最难治疗的类型之一。与此相关的治疗方法采用间接方法,针对 RAS 的下游通路。
RAS 下游通路药物靶点
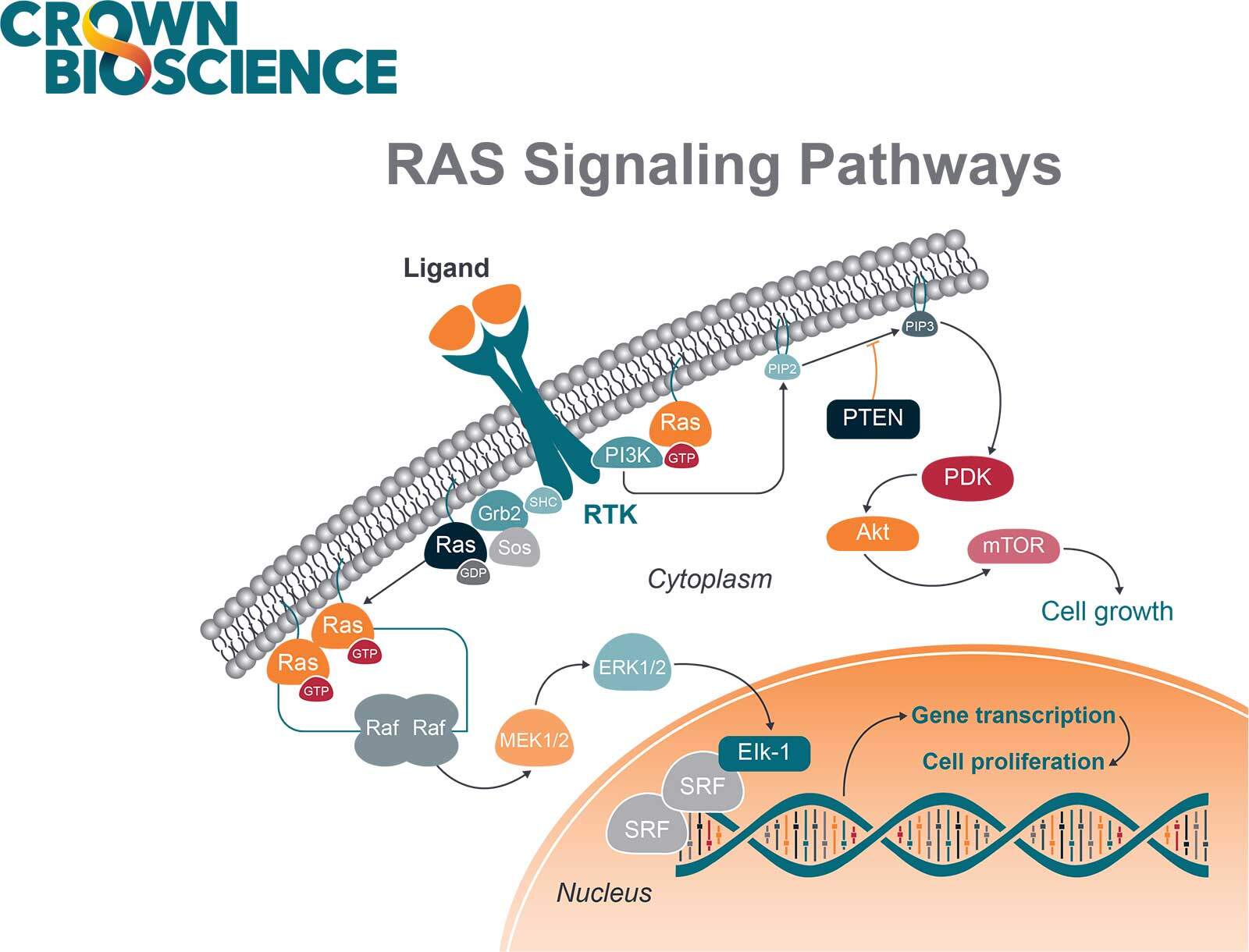
RAS-RAF-MEK-ERK
由配体结合到受体酪氨酸激酶(RTK)从而启动RAS-RAF-MEK-ERK(MAPK)级联反应是一个重要的信号传导通路。一旦激活 RAF,RAF 就会磷酸化并激活丝裂原激活蛋白激酶1和2(MEK1 和 MEK2)。然后这些蛋白可以进一步激活下游的细胞外信号调节激酶1和2(ERK1 和 ERK2)。
ERK1 和 ERK2 通过磷酸化作用于细胞核中的转录因子(例如 c-Myc、c-Fos 和 CREB),调节与细胞生长、细胞分化和分裂相关的基因表达。
KRAS G12C 抑制剂 sotorasib(AMG510)和 adagrasib(MRTX849)分别在 2021 年及 2022 年批准用于 NSCL C治疗,这再次调动了人们开发 KRAS 抑制剂的兴趣。除了 KRAS G12C 突变外,针对其他 KRAS 的特定抑制剂也在开发中。例如,MRTX1133 已被证明以非共价结合形式(KD = ∼0.2 pM)结合 KRAS G12D,其结合选择性比 KRAS WT 高出700倍。TKR15 能够结合 KRAS G12V 蛋白,并以 IC50 为 0.21 µM显著抑制 A549 细胞的增殖。已有研究表明一系列不可逆结合 KRASG12S 并抑制其致癌信号而不会影响 KMS20 细胞中其野生型对应物的小分子。最近,BI-2865 作为泛 KRAS 抑制剂,能够阻止激活野生型 KRAS,并针对包括 G12A/C/D/F/V/S,G13C/D,V14I,L19F,Q22K,D33E,Q61H,K117N 和 A146V/T 在内的广泛 KRAS 突变体。此外,BRAF 抑制剂 vemurafenib 和 dabrafenib 以及 MEK 抑制剂 trametinib 和 cobimetinib 已获批准可用于 BRAF V600E/K 转移性黑色素瘤。
PI3K-Akt
RAS 还与 PI3K-Akt 通路相互作用并刺激该通路。一旦 PI3K 被 RAS 激活,进而AKT被磷酸化并激活,产生多种下游效应。这些效应包括激活 mTOR,mTOR 调控细胞生长、增殖和血管生成所需的蛋白质合成。
PI3K 通路也因其在多种癌症中的过度激活以及对癌细胞增殖和存活的重要性而被选为癌症治疗的靶点。然而,在治疗过程中,发现了诸如反馈激活异常、补偿激活、药物抗性以及 PI3K 通路抑制剂的毒性等问题。
当前抑制途径:BRAF 和 MEK
使用 BRAF 抑制剂(例如 vemurafenib、dabrafenib)和/或 MEK 抑制剂(例如 trametinib、cobimetinib)靶向 MAPK 信号传导已被证明是治疗多种不同癌症的有效方法。然而,抗药性仍是一个主要挑战,大约 30% 的肿瘤对这些抑制剂没有反应,而且在有反应的70%肿瘤中通常也会观察到疾病发展。
RAS 靶向治疗的临床前模型
有多种临床前模型可用于评估针对 RAS 及其下游通路的药剂,包括基因工程化的人源细胞系衍生异种移植模型(CDX)、基因工程小鼠模型(GEMM)以及来源于患者的异种移植模型(PDX)。
基因工程细胞系衍生的小鼠模型
在临床前阶段,人类细胞系可以通过基因突变进行遗传修饰,并利用期建立人类 CDX 模型。
生成原发性或继发性 KRAS 突变的肿瘤细胞系并用于测试针对 RAS 癌基因家族的疗法。这些 CDX 模型已成功用于评估作为单一药物或组合药物使用的 KRAS、BRAF、MEK 和 mTOR 抑制剂的疗效。
例如,携带 KRAS G13D 突变的 HCT116 细胞系和带有 KRAS G12S 突变的 A549 细胞系,为了解 KRAS 驱动的致癌作用的动态以及靶向疗法的潜在疗效提供了宝贵的见解。同样,MiaPaCa-2(KRAS G12C)和 SW48(KRAS G12V)细胞系是评估新型 KRAS 抑制剂的药效学和治疗窗口的关键模型。
这两项研究突出了 KRAS 突变谱系中的复杂性和治疗机会,展示了像 CDX 这样的模型系统在推动临床应用发展中的关键作用。
基因工程小鼠模型
临床前基因工程小鼠模型(GEMM)能够重现疾病特征,模型中与肿瘤进展和发展强烈相关的基因被删除、过度表达或突变。这导致自发性肿瘤形成。
大量可用的 GEMM 涵盖了许多常见的癌症指征,如肺癌、前列腺癌、乳腺癌、结肠癌和胰腺癌。
人们对利用 KRAS 突变型 GEMM 测试针对RAS癌基因家族的疗法特别感兴趣。实际上,在肺、胰腺和胃肠道中条件性表达KRAS突变体能诱导癌前上皮增生、腺瘤、胰腺上皮内瘤变和腺癌。这些模型已成功用于证明 BRAF、MEK 和 mTOR 抑制剂作为单一药物和组合使用的有效性。
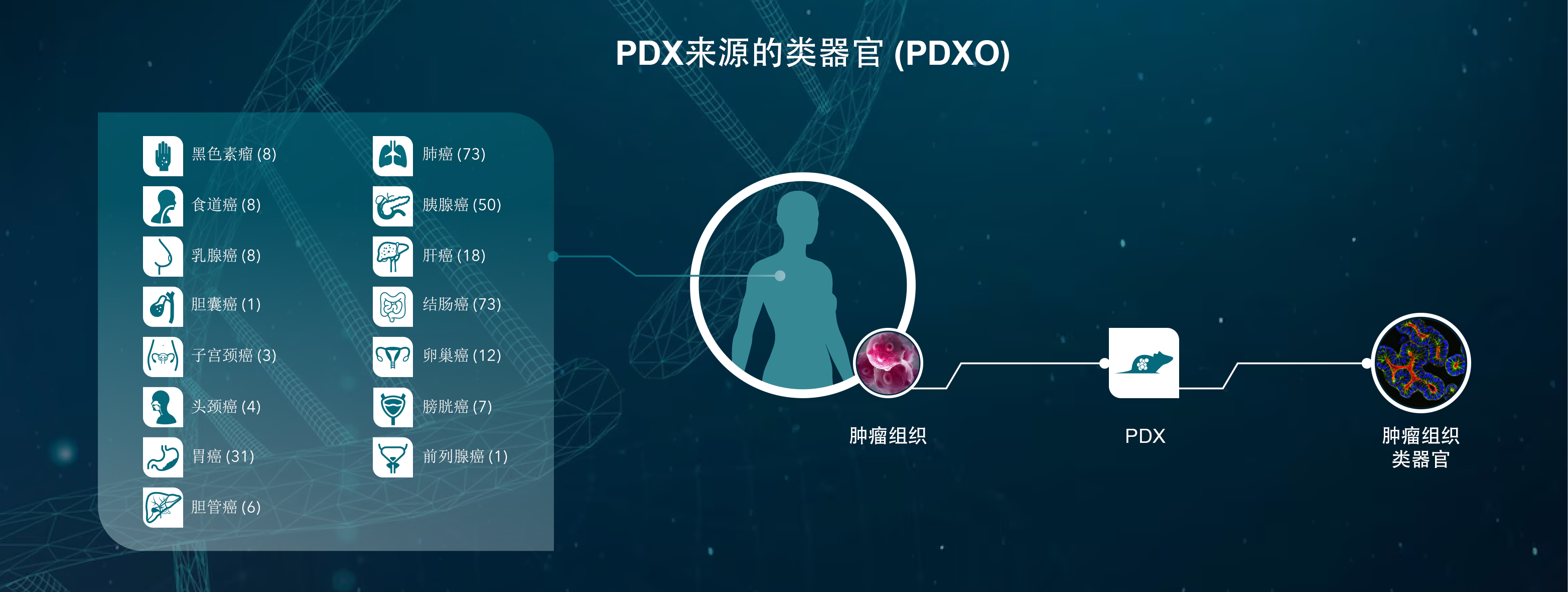
患者来源的异种移植模型
PDX 也是评估 RAS 靶向疗法的有价值且具有转化性的临床前模型。PDX 是从患者组织样本直接衍生的动物模型,它们保持了与来源患者的基因型和表型的保真性。这种模型比传统异种移植更具预测性。
由于它们在时间上接近患者,并且从未受到细胞培养的选择压力,PDX模型能够真实重现患者疾病。这些模型不仅在患者癌症的组织学表现上显示出高保真度,而且在对化疗、放疗和靶向疗法的反应上也是如此。
携带相关突变的PDX模型已成功用于评估 RAS 下游靶点抑制剂的疗效,并且最近用于识别与 KRAS 结合的小分子化合物。
总结
RAS 蛋白是控制细胞生长、增殖和迁移信号的重要组成部分。RAS 突变在人类肿瘤中频繁发现,并且很大程度上对靶向疗法有抵抗性。
基因工程小鼠模型(GEMM)和患者衍生的异种移植模型(PDX)中的 RAS 突变体重现了人类疾病的关键方面,为肿瘤学研究提供了重要的模型,并广泛用于临床前目标和疗法的验证。